БИОХИМИЯ УЧЕБНИК ДЛЯ ВУЗОВ - Е. С. Северина - 2004
РАЗДЕЛ 4. БИОСИНТЕЗ НУКЛЕИНОВЫХ КИСЛОТ И БЕЛКОВ (МАТРИЧНЫЕ БИОСИНТЕЗЫ). ОСНОВЫ МОЛЕКУЛЯРНОЙ ГЕНЕТИКИ
VIII. Механизмы генетической изменчивости. Полиморфизм белков. Наследственные болезни
Точная работа всех матричных биосинтезов — репликации, транскрипции и трансляции — обеспечивает копирование генома и воспроизведение фенотипических характеристик организма в поколениях, т. е. наследственности. Однако биологическая эволюция и естественный отбор возможны только при наличии генетической изменчивости. Установлено, что геном постоянно претерпевает разнообразные изменения. Несмотря на эффективность механизмов коррекции и репарации ДНК, часть повреждений или ошибок в ДНК остаётся. Изменения в последовательности пуриновых или пиримидиновых оснований в гене, не исправленные ферментами репарации, получили название «мутации». Одни из них остаются в соматических клетках, в которых они возникли, а другие обнаруживаются в половых клетках, передаются по наследству и могут проявляться в фенотипе потомства как наследственная болезнь.
Существенный вклад в генетическую изменчивость вносят перестройки хромосом в процессе мейоза. Как уже указывалось ранее, слияние яйцеклетки со сперматозоидом у эукариотов сопровождается генетическими рекомбинациями, в ходе которых происходит обмен участками ДНК между гомологичными хромосомами. Это приводит к появлению потомства с новой комбинацией генов.
Ген или части генов могут перемещаться из одного места хромосомы в другие. Эти подвижные элементы или фрагменты ДНК получили название транспозонов и ретротранспозонов.
Транспозоны — участки ДНК, удаляемые из одного локуса хромосомы и встраиваемые в другой локус той же или другой хромосомы. Ретротранспозоны не покидают исходного положения в молекуле ДНК, но могут копироваться, и копии встраиваются, подобно транспозонам, в новый участок. Включаясь в гены или участки около генов, они могут вызывать мутации и изменять их экспрессию.
Геном эукариотов подвергается изменениям и при заражении ДНК- или РНК-содержащими вирусами, которые внедряют свой генетический материал в ДНК клеток хозяина.
А. Мутагенез
Изменения в геноме могут быть разнообразны и затрагивать различные по протяжённости участки ДНК от хромосом и генов до отдельных нуклеотидов (табл. 4-7).
Таблица 4-7. Классификация мутаций
Тип мутаций |
Характер мутационных изменений |
Примеры последствий |
Геномный |
Изменение числа хромосом |
Болезнь Дауна (появление дополнительной хромосомы 21) |
Хромосомные |
Общее число хромосом не меняется. Наблюдают перестройки хромосом, обычно видимые при микроскопическом исследовании. |
Мышечная дистрофия Дюшенна (делеции Х-хромосомы) |
Генные |
Изменения затрагивают один кодон или небольшой отрезок гена и не обнаруживаются цитогенетически |
Серповидно-клеточная анемия, вызванная заменой одного нуклеотида в гене β-цепи глобина |
Наиболее драматичны геномные и хромосомные мутации, часто наблюдаемые на уровне соматических клеток. Если они имеют место в половых клетках, то для организма это имеет чаще всего летальные последствия. Частота мутаций в половых клетках высока. Существуют данные, указывающие на то, что в 20% случаев при беременности у эмбрионов наблюдают нарушения структуры хромосом. В 90% случаев это приводит к ненормальному развитию плода и элиминированию зародышей в результате спонтанных абортов. Выкидыши, происходящие в течение первых нескольких недель беременности, связаны с серьёзными нарушениями хромосом. В 50% случаев отмечается трисомия по аутосомам, т. е. вместо пары хромосом наблюдаются три. Пример такой патологии — болезнь Дауна, при которой хромосома 21 присутствует в 3 экземплярах.
Некоторые генные мутации закрепляются в популяции, становятся наследственными и определяют эволюционные процессы. С мутациями такого типа связано появление различных наследственных патологий, сопровождающихся прекращением синтеза белка, кодируемого Повреждённым геном, либо синтезом изменённого белка.
Генные, или точечные, мутации бывают в основном 3 видов: замены, при которых одно азотистое основание в ДНК замещается на другое; вставки, обеспечивающие внедрение в молекулу ДНК одного или нескольких дополнительных нуклеотидов; делении (или выпадения) одного или нескольких нуклеотидов, при которых происходит укорочение молекулы ДНК.
1. Мутации по типу замены возникают в результате замены одного азотистого основания на другое, что вызывает изменение в одном из кодонов мутантного гена. Если кодирующий триплет, в котором находится изменённый нуклеотид, из-за вырожденности кода вызывает включение в белок той же аминокислоты, что исходный кодон (или кодон «дикого» типа), то такую мутацию называют «молчащей», и белковый продукт остаётся тем же.
Когда замена одного основания приводит к замене аминокислоты в мутантном белке, то такую мутацию называют «миссенс-мутация». В ряде случаев, несмотря на произошедшую замену, белок сохраняет биологическую активность. Это, как правило, связано с тем, что изменённая аминокислота находится в участке белка, не имеющем функционального значения, и к тому же она по структуре и свойствам напоминает исходную аминокислоту. Такая мутация тоже будет «молчащей», а замена — эквивалентной.
Триплет «дикого» типа |
Изменённый триплет |
|
Матрица ДНК |
3'-ТАА-5' |
3'-GАА-5' |
Кодон мРНК |
5'-АUU-3' |
5'-СUU-3' |
Аминокислота |
-Иле- |
-Лей- |
Иногда аминокислота, оказавшаяся заменённой, располагается в области, важной для проявления функциональной активности белка, и её замещение приводит к образованию функционально неактивного продукта. Так, точечная мутация в кодоне серина (Сер — важнейший структурный компонент активного центра сериновых протеаз: трипсина, химотрипсина и некоторых других ферментов) приводит к полной потере активности. Если подобный фермент участвует в реакциях главных метаболических путей, то такая «неэквивалентная» замена может стать летальной.
Триплет «дикого» типа |
Изменённый триплет |
Триплет «дикого» типа |
Изменённый триплет |
||
Матрица ДНК |
3'-GGТ-5' |
3'-GGА-5' |
Матрица ДНК |
3'-АGА-5' |
3'-ААА-5' |
Кодон мРНК |
5'-ССА-3' |
5'-ССU-3' |
Кодон мРНК |
5'-UCU-3' |
5'-UUU-3' |
Аминокислота |
-Про- |
-Про- |
Аминокислота |
-Сер- |
-Фен- |
В ряде случаев мутантный белок, несмотря на входящую в него изменённую аминокислоту, сохраняет способность выполнять свою функцию, но может быть не столь эффективным, как белок «дикого» типа. В результате мутации у фермента может оказаться более высоким значение Кm или более низким значение Vmах, а иногда то и другое одновременно. Такие частично функционирующие белки называют мутантными белками с неполностью подавленной функцией.
Изредка в результате мутации белковый продукт гена оказывается лучше приспособленным к выполнению своей функции. Такие мутации дают потомству преимущества в борьбе за существование, а серия соответствующих мутаций может привести к появлению нового вида.
Наибольшим повреждающим действием обладают мутации, приводящие к образованию одного из терминирующих кодонов (нонсенс- мутация). В процессе синтеза белка работа рибосомы будет остановлена на мутантном триплете мРНК: UАА, UАG или UGА. Проявление нонсенс-мутаций зависит от их внутригенной локализации. Чем ближе мутация к 5'-концу гена, т. е. к началу транскрипции, тем короче её белковый продукт, а, следовательно, тем меньше он способен к осуществлению биологической функции.
Триплет «дикого» типа |
Изменённый триплет |
|
Матрица ДНК |
3'-GТС-5' |
3'-АТС-5' |
Кодон мРНК |
5'-САG-3' |
5'-UАG-3' |
Аминокислота |
-Глн- |
Стоп-кодон |
2. Мутации по типу вставки или делеции нуклеотидов
Более многочисленны и опасны для клеток мутации по типу вставки или делеции (утраты) нуклеотидов.
Если мутация приводит к вставке или делеции в ген одной нуклеотидной пары или участка двухцепочечной молекулы ДНК с числом мономеров, не кратным 3, то это вызывает изменение считывания всех последующих кодонов, так как происходит сдвиг «рамки считывания» ДНК и нарушение соответствия между кодонами в ДНК и аминокислотами в конечном продукте — белке (рис. 4-57).
Рис. 4-57. Делеции (А) или вставки (Б) нуклеотидов вызывают мутации со сдвигом «рамки считывания».

Как видно из рис. 4-57, нарушения в прочтении информации начинаются с участка, в котором произошла мутация, так как именно в этом месте происходит сдвиг «рамки считывания» информации. Белковый продукт за точкой мутации будет иметь случайную последовательность аминокислот. Мутации со сдвигом рамки считывания часто приводят к появлению внутреннего терминирующего кодона, вызывающего преждевременное прекращение синтеза полипептидной цепи и образование укороченного продукта, лишённого биологической активности.
Мутации со сдвигом «рамки считывания» индуцируют ингибиторы матричных синтезов — «интеркаляторы». Их большие плоские молекулы, похожие на обычные азотистые основания или пары оснований, встраиваются между двумя соседними парами оснований, в результате в ДНК «как бы» появляется лишнее основание. В ходе репликации такой изменённой цепи ДНК в дочернюю нить в результате ошибочного спаривания с «интеркалированной» молекулой может встроиться дополнительный нуклеотид.
Иногда, хотя и крайне редко, теряется или включается в ДНК олигодезоксинуклеотид, состоящий из 3 или кратного 3 числа нуклеотидов. Такие мутации называют делениями или вставками без сдвига «рамки считывания» ДНК. В образующемся белковом продукте в этом участке окажется пропущенной или, наоборот, включённой дополнительно одна или несколько аминокислот, тогда как вся остальная аминокислотная последовательность будет соответствовать исходной молекуле. Такие мутации, как правило, не приносят большого вреда.
Информация о разных типах мутаций и изменениях в структуре мутантных белков обобщены в табл. 4-8.
Таблица 4-8. Основные виды генных мутаций
Виды мутаций |
Изменения в структуре ДНК |
Изменения в структуре белка |
Замена Без изменения смысла кодона С изменением смысла кодона (миссенс-мутация) С образованием терминирующего кодона (нонсенс-мутация) |
Замена одного нуклеотида в кодоне |
Белок не изменён Происходит замена одной аминокислоты на другую Синтез пептидной цепи прерывается, и образуется укороченный продукт |
Вставка Без сдвига «рамки считывания» Со сдвигом «рамки считывания» |
Вставка фрагмента ДНК из 3 нуклеотидов или с числом нуклеотидов, кратным 3 Вставка одного или нескольких нуклеотидов, не кратных 3 |
Происходит удлинение полипептидной цепи на одну или несколько аминокислот Синтезируется пептид со «случайной» последовательностью аминокислот, так как изменяется смысл всех кодонов, следующих за местом мутации |
Делеция Без сдвига «рамки считывания» Со сдвигом «рамки считывания» |
Выпадение фрагмента ДНК из 3 нуклеотидов или с числом нуклеотидов, кратным 3 Выпадение одного или нескольких нуклеотидов, не кратных 3 |
Происходит укорочение белка на одну или несколько аминокислот Синтезируется пептид со «случайной» последовательностью аминокислот, так как изменяется смысл всех кодонов, следующих за местом мутации |
3. Частота мутаций
Считается, что средняя частота возникновения мутаций в структурных локусах (областях локализации гена в хромосоме или в молекуле ДНК) человека колеблется в пределах от 10-5 до 10-6 на одну гамету за каждое поколение. Однако эта величина может значительно варьировать для разных генов (от 10-4 для генов с высокой скоростью мутаций до 10-11 для наиболее устойчивых участков генома). Столь существенные колебания в частоте возникновения мутаций обусловлены характером мутационного повреждения, механизмом возникновения мутации, протяжённостью кодирующей области мутантного гена, функциями белка, закодированного в этом гене. Так, для гена гемоглобина скорость замещения одного основания другим лежит в интервале μ = 2,5 x 10-9 — 5 x 10-9 замен в гамете за одно поколение. Чтобы представить себе, что означают эти цифры, распространим эту скорость мутаций на весь геном человека — 3 x 109 пар оснований. Умножив размер генома на скорость μ, мы получим, что геном за одно поколение может получить от 7 до 15 мутаций, т. е. это значит, что каждая гамета содержит такое количество изменений в ДНК по сравнению с родительской ДНК. А поскольку у каждого индивидуума клетки диплоидны и получаются при слиянии 2 гамет, то мутаций тоже в 2 раза больше.
Спрашивается, каким же образом человечество справляется с такой мутационной нагрузкой? Отвечая на этот вопрос, следует помнить, что кодирующие части генов, изменения в которых наиболее опасны, занимают не более 10% генома. Ситуация облегчается ещё и тем, что далеко не каждая мутация в кодирующей области имеет фенотипическое проявление. Многие попадают в 3'-положение кодонов и, таким образом, являются «молчащими», так как благодаря вырожденности генетического кода они не приводят к аминокислотным заменам, другие оказываются в доменах, несущественных для функционирования белков. Потомству передаются мутации, происходящие в гаметах, а их процент совсем невелик.
Б. Организация генома человека
Геном эукариотических клеток (клеток человека в частности) содержит значительно больше ДНК, чем геном прокариотов. У кишечной палочки Е. соli ДНК содержит 3,8 x 106 пар нуклеотидов или около 3000 генов. При этом вся ДНК выполняет определённые функции: кодирует белки, рРНК, тРНК или участвует в регуляции образования генных продуктов. Общая длина ДНК гаплоидного набора из 23 хромосом человека составляет 3,5 x 109 пар нуклеотидов, что в 1000 раз превосходит размер генома прокариотов. Этого количества ДНК достаточно для создания нескольких миллионов генов. Однако, согласно многим независимым подсчётам, истинное число структурных генов находится в области 100 000, а по данным Международного консорциума и фирмы «Целера Геномикс», полученным при секвенировании генома человека и опубликованным в 2001 г., число белок-кодирующих генов, по оценкам первой организации, составляет 31780, а по оценкам второй — 39 114 генов. Последовательность нуклеотидов в ДНК человека имеет области, кодирующие белки (не более 2% общего генома), области, кодирующие РНК (около 20% генома), и повторяющиеся последовательности (более 50% общего генома).
Функции «избыточной» ДНК до конца не ясны, полагают, что она участвует в регуляции экспрессии генов, процессинга РНК, выполняет структурные функции, повышает точность гомологичного спаривания и рекомбинации хромосом в процессе мейоза, способствует успешной репликации. Большая часть этой ДНК возникла в результате обратной транскрипции РНК и благодаря наличию подвижных элементов.
Часто повторяющиеся последовательности ДНК
Во фракции «избыточной» ДНК выделяют относительно короткие последовательности длиной от 2 до 10 пар нуклеотидов, повторяющиеся миллионы раз. Эти часто повторяющиеся последовательности получили название «сателлитной ДНК». Они составляют около 10% всего генома человека, рассеяны по всему генетическому материалу клетки, но преимущественно локализуются в центромерных и теломерных областях большинства хромосом.
Умеренно повторяющиеся последовательности ДНК
Выделяют также группу умеренно повторяющихся последовательностей ДНК, очень гетерогенную по длине и числу копий, составляющую более 30% генома человека. Эта фракция включает ДНК, кодирующую структуру рРНК, тРНК и некоторых мРНК. Гены гистонов присутствуют в геноме в количестве нескольких сотен копий и также принадлежат к этому классу. Умеренно повторяющиеся последовательности ДНК включают участки, которые не транскрибируются, но важны в регуляции экспрессии генов: промоторы и энхансеры. В этой группе были обнаружены последовательности ДНК длиной примерно в 300 пар нуклеотидов, которые повторяются около миллиона раз (в среднем через каждые 5000 пар нуклеотидов) и рассеяны по всему геному человека.
Частые представители умеренно повторяющихся последовательностей — line-семейство (от англ. long interspersed elements), или семейство длинных рассеянных элементов, в которое входят ДНК последовательности длиной от 5000 — 8000 пар нуклеотидов, встречаются в геноме в количестве 850 000 копий.
Уникальные последовательности ДНК
Они представлены ДНК-последовательностями, которые присутствуют в геноме в количестве одной или нескольких копий, и транскрибируются, образуя мРНК, содержащие информацию о различных белках (рис. 4-58).
Рис. 4-58. Распределение уникальных, умеренно и часто повторяющихся последовательностей ДНК в гипотетической хромосоме человека. Уникальные последовательности (3) транскрибируются в виде мРНК. Эти гены встречаются в одной или нескольких копиях. Гены рРНК и тРНК представлены множеством копий, образующих кластеры в геноме. Гены большой пре-рРНК образуют ядрышковый организатор. Умеренно повторяющиеся последовательности (2) распределены по всему геному, а часто повторяющиеся последовательности (1) образуют кластеры в областях центромеры и концов хромосомы — теломеров.

Типичный ген человека состоит примерно из 28 000 нуклеотидов и содержит в среднем 8 экзонов, его кодирующая последовательность составляет около 1340 пар нуклеотидов и шифрует белок, включающий 447 аминокислотных остатков. Самый крупный ген — ген мышечного белка дистрофина, состоящий из 2,4 х 106 пар нуклеотидов, а наибольшее количество экзонов (234) присутствует в гене фибриллярного белка титина, ответственного за пассивную эластичность скелетных мышц.
Нередко уникальные последовательности образуют мультигенные семейства, располагающиеся в виде кластеров в определённых областях одной или нескольких хромосом. Примерами мультигенных семейств могут служить гены рибосомальных, транспортных и малых ядерных РНК, гены α- и β-глобинов, тубулинов, миоглобина, актина, трансферина и многих других.
В мультигенных семействах наряду с функционально активными генами содержатся псевдогены — мутационно изменённые последовательности, не способные транскрибироваться или продуцирующие функционально неактивные генные продукты.
Псевдогены — одна из важных структурных особенностей генома человека. Эти уникальные последовательности очень сходны по структуре с определёнными генами. В связи с тем, что в генных семействах наряду с псевдогенами сохраняются неизменённые гены, жизнеспособность организма не нарушается. Для многих генов обнаружены псевдогены. Их количество варьирует от одной до нескольких десятков копий на геном, и, как правило, они расположены тандемно. Иногда псевдогены и соответствующие им нормальные гены локализованы в разных хромосомах.
В. Полиморфизм белков
Поскольку большинство нормальных клеток человека диплоидны, то они содержат две копии каждой хромосомы, одна из которых получена от отца, а вторая от матери. Эти две копии одной и той же хромосомы называют гомологичными (рис. 4-59). В ДНК каждой хромосомы содержится более тысячи генов. Соответствующие друг другу гены в гомологичных хромосомах называют аллелями. Аллели могут быть идентичными и содержат одинаковую последовательность нуклеотидов. В этом случае индивидуум, имеющий такие аллели, будет гомозиготен по данному признаку. Если аллели различаются по последовательности нуклеотидов в ДНК, то говорят о гетерозиготном наследовании гена. В этом случае индивидуум будет иметь 2 белковых продукта гена, различающихся по аминокислотной последовательности.
У каждого человека существует только 2 разных аллеля одного гена, тогда как в популяции людей вариантов аллелей может быть огромное множество. Как уже говорилось ранее, изменчивость структуры ДНК, а, следовательно, разнообразие аллелей, обусловлено мутационным процессом и рекомбинациями в гомологичных хромосомах половых клеток. Если в ходе мейоза рекомбинации сопровождаются обменом участками ДНК, меньшими по размеру, чем ген, то такой процесс может приводить к появлению новых, прежде не существовавших аллелей. А поскольку рекомбинации — более частые события, чем мутации в кодирующих участках гена, то разнообразие вариантов аллелей обусловлено главным образом ими.
Рис. 4-59. Гомологичные хромосомы и соответствующие аллелям белковые продукты. На рисунке показано расположение 4 аллелей (АА, Вb, СС, dD) на гомологичных хромосомах. Аллели могут быть идентичны, как в случае генов АА и СС, или различаться (Вb, Dd). Белковые продукты будут идентичны для аллелей АА и СС, но будут различаться по аминокислотной последовательности в случае аллелей Вb и Dd.

Существование в популяции 2 и большего числа аллелей одного гена называют «аллеломорфизм», или «полиморфизм», а белковые продукты, образующиеся в ходе экспрессии этих вариантов гена — «полиморфы». Разные аллели встречаются в популяции с разной частотой. К полиморфам относят только те варианты, распространённость которых в популяции не меньше 1%.
В процессе эволюции отдельные гены амплифицируют с образованием копий, а их структура и положение могут изменяться в результате мутаций и перемещений не только внутри хромосомы, но и между хромосомами. Со временем это приводит к появлению новых генов, кодирующих белки, родственные исходному, но отличающиеся от него определёнными свойствами и занимающие в хромосомах разные генные локусы (или места).
К родственным белкам относят изобелки, представляющие собой варианты белков, выполняющие одну и ту же функцию и обнаруживаемые в пределах одного вида организмов. Так, в группе из 2000 генов человека, кодирующих факторы транскрипции и транскрипционные активаторы, идентифицировано 900, относящихся к семейству белков, имеющих «цинковые пальцы». Существует 46 генов фермента глицеральдегид- 3-фосфатдегидрогеназы, осуществляющего единственную окислительную реакцию в метаболическом пути катаболизма глюкозы до пирувата.
Выявлены семейства родственных белков, возникшие в ходе эволюции из одного «предкового» гена, или гена-предшественника. Такие семейства составляют:
✵ гены миоглобина и протомеров гемоглобинов;
✵ группа протеолитических ферментов: трипсин, химотрипсин, эластаза, плазмин, тромбин и некоторые другие белки и ферменты.
1. Гемоглобины человека
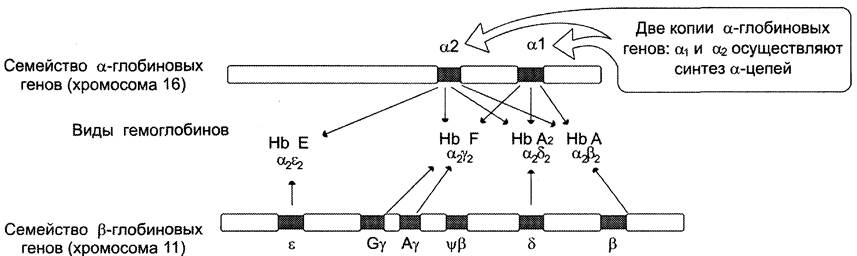
В ходе эволюции из единичных генов-предшественников возникли семейства генов α- и β-глобинов (рис. 4-60), на хромосомах 16 и 11 соответственно.
В процессе онтогенеза у людей образуются разные виды гемоглобинов, обеспечивающие наилучшую адаптацию к меняющимся условиям существования. НbЕ — эмбриональный, синтезируется у зародыша в первые месяцы развития, НbЕ — фетальный, обеспечивает дальнейшее внутриутробное развитие плода, а НbА и НbА2осуществляют транспорт кислорода в организме взрослого человека. Эти белки представляют собой тетрамеры, состоящие из полипептидных цепей двух видов: α и β в НbА (2α2β), α и ε в НbЕ (2α2ε), а у остальных гемоглобинов β-цепи заменены на y-полипептиды в НbF (2α2y) или на 5-цепи в НbА2 (2α2δ).
Рисунок 4-60. Схематическое расположение кластеров α- и β-глобиновых генов. Обнаружены две копии α-глобинового гена: α1 и α2, каждая из которых обеспечивает синтез α-глобиновой цепи. В семействе генов β-глобинов локус ε экспрессируется в период раннего эмбрионального развития плода на начальных месяцах беременности (α2ε2). Гены у экспрессируются в процессе внутриутробного развития плода (НbF, α2y2). Гемоглобины взрослого человека — НbА (аΨ2), составляющий 96%, и НbА2 (α2δ2), составляющий 2-2,5%, образуются в результате экспрессии генов β и δ. Ψβ — псевдоген, имеющий последовательность нуклеотидов, гомологичную β-гену, но содержащий мутации, нарушающие его экспрессию.
Полиморфизм гемоглобинов в популяции людей очень велик. Наряду с генами, кодирующими изобелки и занимающими разные локусы на хромосоме, обнаружено большое число вариантов гемоглобина А, являющихся продуктами аллельных генов. Некоторые варианты НbА представлены в таблице 4-9.
Таблица 4-9. Некоторые варианты гемоглобина А человека
Название |
Мутация. |
Аномальное свойство |
НbI |
Замены аминокислот α16Лиз —> Глу |
Нет |
Torino |
α43Фен —> Вал |
Нарушен контакт с гемом; нестабилен |
Nasharon |
α47Асп —> Гис |
Нестабилен |
Buda |
α61Лиз —> Асн |
Снижено сродство к O2 |
Iwate |
α87Гис —> Тир |
Снижено сродство к O2; легко окисляется в МеtHb |
Denmark Hill |
а95Про —>Ала |
Нарушен α1β2-контакт; сродство к O2 повышено |
HbC |
β6Глу —>Лиз |
Нет |
HbS |
β6Глу —> Вал |
Снижены растворимость и сродство к O2 |
Baltimore |
β16Глу —> Асн |
Нет |
Genova |
β28Лей —> Про |
Повышено сродство к O2 |
Zurich |
β6ЗГис —> Арг |
Повышено сродство к O2; нестабилен |
Koln |
β98Вал —> Мет |
То же |
Kansas |
β102Асн —> Тре |
Нарушен α1β2-контакт; сродство к O2 повышено |
San Diego |
β109Вал —> Мет |
Нарушен α1β1-контакт; сродство к O2 снижено |
Hiroshima |
β146Гис —> Асп |
Сильно повышено сродство к O2 |
Leiden |
Делеции аминокислот β6 или β7 —> 0 |
Нестабилен |
Tochigi |
β(56-59) —> 0 |
Нестабилен |
Green Hill |
β(91-95) —> 0 |
Нестабилен, повышено сродство к O2 |
Tak |
Вставки аминокислот Цепь удлинена на 10 остатков с С-конца |
Повышено сродство к O2 |
Примечание. Цитировано по учебнику А. Я. Николаева «Биологическая химия» (М.: Высшая школа, 1989).
Один из наиболее известных аллельных вариантов НbА — НbS, образующийся в результате замены остатка глутамата в положении 6 β-цепи НbА на валин (β6 Глу —> Вал). По аллелям НbА и НbS всех людей можно разделить на 3 генотипически различающиеся группы: АА, АS и SS. Распространённость аллеля Б по земному шару неравномерна. Часто людей с этим аллелем можно встретить в малярийных районах Африки и Азии (до 35%). К настоящему времени описано свыше 300 вариантов НbА, на основании этого признака всех людей можно разделить на 600 генотипических групп по наиболее часто встречающимся аллелям.
2. Группы крови
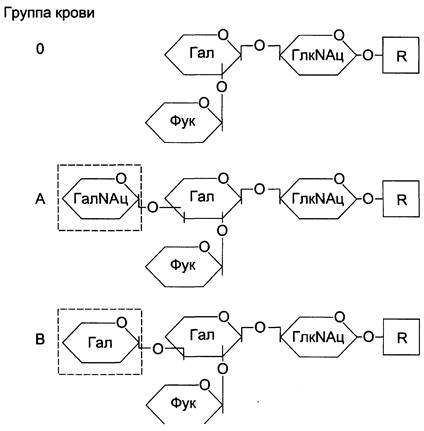
Другой важный пример полиморфизма белков, связанный с проблемой переливания крови, — существование в популяции людей 3 аллельных вариантов гена фермента гликозил- трансферазы (А, В и 0). Этот фермент принимает участие в синтезе олигосахарида, локализованного на наружной поверхности плазматической мембраны и определяющего антигенные свойства эритроцитов. Варианты фермента А и В имеют разную субстратную специфичность: вариант А катализирует присоединение к олигосахариду N-ацетилгалактозамина, а вариант В — галактозы. Вариант 0 кодирует белок, лишённый ферментативной активности. В результате структура олигосахаридов, расположенных на поверхности эритроцитов, будет разной (рис. 4-61).
Рис. 4-61. Структура олигосахаридов, определяющих группу крови. Олигосахариды различаются концевыми мономерами. Олигосахарид А имеет на нередуцирующем конце N-ацетилгалактозамин (ГалNАц), олигосахарид В — галактозу (Гал), а олигосахарид 0 укорочен на один моносахаридный остаток. R представляет собой белок либо липид — церамид.
Антитела к антигенам А и В обычно имеются в сыворотке крови людей, на поверхности эритроцитов которых отсутствует соответствующий антиген, т. е. индивидуумы с антигенами А на поверхности эритроцитов продуцируют в сыворотку крови антитела к В-антигенам (анти-В), а люди с В-антигенами — антитела к антигенам А (анти- А). В сыворотке крови анти-А и анти-В обычно присутствуют в высоких титрах и при появлении соответствующих антигенов способны активировать ферменты системы комплемента.
При переливании крови руководствуются правилом, согласно которому кровь донора и реципиента не должна содержать антигены и антитела, реагирующие между собой: например, реципиенту, имеющему в сыворотке крови анти- А, нельзя переливать кровь от донора, содержащего на эритроцитах антигены А.
При нарушении этого правила происходит реакция антиген-антитело. Это вызывает агглютинацию (склеивание) эритроцитов и их разрушение ферментами комплемента и фагоцитами.
Как видно из табл. 4-10, у индивидуумов-гетерозигот, имеющих группу крови АВ (IV), на эритроцитах присутствуют А- и В-антигены, функционируют 2 варианта гликозилтрансферазы (А и В), а следовательно антитела не образуются. Этих людей можно рассматривать как «универсальных» реципиентов, которым безопасно вводить эритроциты от доноров, имеющих любые группы крови. Однако люди с группой крови IV не могут безопасно получать сыворотку крови от этих доноров, так как она содержит антитела к А- и/или В-антигенам. В то же время индивидуумы, имеющие 0 (I) группу крови, — гомозиготы по неактивному варианту гликозилтрансферазы 0, и поверхность их эритроцитов лишена антигенов. Такие люди являются «универсальными» донорами эритроцитарной массы, так как их эритроциты можно вводить людям с группами крови А, В, 0 или АВ. В то же время сыворотка крови этих доноров содержит антитела к А- и В-антигенам и может использоваться только для пациентов 0 (I) группы крови.
Таблица 4-10. Характеристика групп крови
Антигены эритроцитов |
Нет |
А |
В |
АВ |
Г енотипы |
00 |
АА или АО |
ВВ или В0 |
АВ |
Антитела в сыворотке крови |
Анти-А и анти-В |
Анти-В |
Анти-А |
Нет |
Группы крови |
0 (I) |
А (II) |
В (III) |
АВ (IV) |
Частота (%) |
45 |
40 |
10 |
5 |
3. Белки главного комплекса гистосовместимости и трансплантационная несовместимость
При формировании клеточного иммунного ответа узнавание Т-лимфоцитами чужеродного антигена происходит только если он расположен рядом с гликопротеинами, присутствующими на собственной клеточной мембране. Эти гликопротеины называют белками главного комплекса гистосовместимости, или МНС-белками (см. раздел 1). Существуют 2 класса этих белков: молекулы класса I и II. МНС-белки класса I обнаружены практически во всех содержащих ядро клетках, включая Т- киллеры, тогда как МНС-белки класса II найдены главным образом в клетках, участвующих в иммунном ответе, в антиген-представляющих В- клетках и Т-хелперах, но не в Т-киллерах и макрофагах.
Строение МНС-белков кодирует семейство генов, расположенных на коротком плече хромосомы 6 и занимающих участок ДНК длиной более 6000 пар нуклеотидов. Это семейство состоит из серии тесно сцепленных генов, ответственных за синтез МНС-белков и некоторых компонентов системы комплемента. Гены комплекса отличаются чрезвычайно высоким полиморфизмом. Число разных аллелей достигает нескольких миллионов. Белки МНС-системы считают самой полиморфной системой человека. Вариабельность МНС-белков обеспечивает трансплантационную несовместимость. Клетки трансплантата имеют набор этих белков, отличный от МНС-белков реципиента (во всех случаях, кроме генетически идентичных близнецов), и это приводит к развитию реакции клеточного иммунитета, в результате которой трансплантированная ткань отторгается.
Исследования показали, что полиморфизм различных белков настолько велик, что можно говорить о биохимической индивидуальности и уникальности каждого человека.
Г. Наследственные болезни
Каждый генетический локус характеризуется определённым уровнем изменчивости, т. е. присутствием различных аллелей у разных индивидуумов. Аллели генов делят на 2 группы — нормальные, или аллели «дикого» типа, для которых функция гена не нарушена, и мутантные, приводящие к нарушению работы гена. «Плохой» аллель кодирует синтез белка, функция которого сильно нарушена и при гомозиготном наследовании фенотипически проявляется как наследственная болезнь. Наследственные болезни — следствие мутаций, произошедших в гаметах или зиготе. Такие мутации могут быть первичными, если возникли в гаметах или в процессе формирования зиготы, или вторичными, если мутантный ген возник раньше и был передан последующему поколению по наследству.
Первичные мутации, как правило, не сопровождаются возникновением болезни, так как происходят обычно в одной из хромосом, и индивидуум, получивший такую мутацию, становится гетерозиготным носителем повреждения в гене. Мутантный ген в гетерозиготном состоянии часто не проявляется как болезнь и существенно не снижает жизнеспособность организма. Это способствует его распространению в популяции.
При вторичных мутациях, если каждый из родителей является носителем мутантного гена, будучи гетерозиготой, возможно рождение детей-гомозигот по дефектному аллелю. В таком случае развивается наследственная болезнь, часто сопровождаемая очень тяжёлым течением.
Согласно данным Всемирной организации здравоохранения, около 2,4% всех новорождённых на земном шаре страдают теми или иными наследственными нарушениями. Около 40% ранней младенческой смертности и инвалидности с детства обусловлены наследственной патологией.
К настоящему времени на хромосомах человека выявлено около 800 генов, мутации в которых приводят к развитию различных наследственных болезней. Количество моногенных заболеваний (т.е. вызванных мутациями в определённом гене) ещё больше и равно примерно 950 в результате существования так называемых «аллельных серий», т. е. групп болезней, клинически сильно отличающихся друг от друга, но обусловленных мутациями в одном и том же гене. Например, мутации в гене рецептора с тирозинкиназной активностью ret могут вызывать 4 различных наследственных заболевания.
Более половины генов, в которых найдены мутации, вызывающие наследственные заболевания, охарактеризованы методами молекулярного анализа. Наибольшую по размеру группу составляют ферменты (31% от общего числа). За этой группой следуют белки, модулирующие функции белков и участвующие в правильном сворачивании полипептидных цепей (14%).
На каждой хромосоме в среднем идентифицировано около 30 структурных генов, мутации в которых вызывают наследственные болезни. Однако распределены эти гены по хромосомам неравномерно. Так, например, на хромосоме 2 их в 3 раза меньше, чем на хромосоме 1. Наибольшее число мутантных генов (более 100) установлено на Х-хромосоме.
Хорошо изученными наследственными заболеваниями, связанными с нарушением синтеза α- или β-цепей Нb, являются талассемии. Синтез α- и β-цепей в норме регулируется таким образом, что все молекулы протомеров используются на синтез тетрамера α2β2. Талассемии возникают как результат мутаций, включающих замены или делеции одного, или нескольких нуклеотидов, а иногда и целого гена, кодирующего структуру одного из протомеров. Эти болезни классифицируют по 4 типам: так, в случае, если одна из цепей не синтезируется, то их обозначают как α0- или β0-талассемии, а если синтез какой-либо из цепей снижен, то α+- или β+-талассемии.
α-Талассемии возникают при нарушении синтеза α-цепей. В геноме каждого индивидуума существует 4 копии гена α-глобина (по 2 копии на каждой хромосоме), поэтому встречаются несколько видов недостаточности α-цепей. Если дефектна одна из 4 копий, то фенотипически это не проявляется, и такого человека рассматривают как «молчащего носителя» талассемии. При дефекте в 2 копиях гена у носителя мутации обнаруживают слабовыраженные признаки болезни, а при дефекте в 3 копиях развивается гемолитическая анемия. При полном отсутствии синтеза α-цепей (т. е. дефектны все 4 копии гена) наступает внутриутробная гибель плода, так как не образуются фетальные формы Нb, а тетрамеры y4 обладают высоким сродством к кислороду и не способны функционировать как транспортные белки.
β-Талассемии развиваются в результате снижения синтеза β-цепей Нb, для которых на каждой хромосоме имеется по одному гену. Синтез НbА начинается после рождения ребёнка. При дефекте в одной из копий гена недостаточность Нb проявляется в слабой степени и не требует специального лечения. Однако при полном выключении синтеза β-цепей развивается тяжёлая форма анемии, и таким пациентам проводят либо периодическую трансфузию крови, либо пересадку костного мозга.
Со многими моногенными наследственными заболеваниями читатель познакомится практически во всех последующих разделах учебника. Здесь же хотелось бы только отметить, что наряду с болезнями, наследственная природа которых ярко выражена, существует множество болезней, характеризующихся семейной предрасположенностью. Это такие широко распространённые заболевания, как сахарный диабет, подагра, атеросклероз, шизофрения и ряд других. В отличие от моногенных болезней, эти заболевания относят к мультифакторным. Поэтому исследования, направленные на выявление белков, аллельные формы которых ответственный за предрасположенность к заболеванию, являются задачами настоящего и будущего времени.