БИОХИМИЯ УЧЕБНИК ДЛЯ ВУЗОВ - Е. С. Северина - 2004
РАЗДЕЛ 7. ОБМЕН УГЛЕВОДОВ
XIII. Метаболизм фруктозы и галактозы
Метаболизм фруктозы и галактозы включает пути использования их для синтеза других веществ (гетерополисахаридов, лактозы и др.) и участие в энергообеспечении организма. В последнем случае фруктоза и галактоза превращаются в печени либо в глюкозу, либо в промежуточные продукты её метаболизма. Таким образом, в результате фруктоза и галактоза наряду с глюкозой могут быть окислены до СO2 и Н2O или использованы на синтез гликогена и триацилглицеролов.
Причиной нарушения метаболизма фруктозы и галактозы может быть дефект ферментов, катализирующих промежуточные реакции их обмена. Эти нарушения встречаются относительно редко, но могут представлять достаточно серьёзную опасность, так как накапливаемые промежуточные метаболиты фруктозы и галактозы обладают токсичностью.
А. Метаболизм фруктозы
Значительное количество фруктозы, образующееся при расщеплении сахарозы, прежде чем поступить в систему воротной вены, превращается в глюкозу уже в клетках кишечника. Другая часть фруктозы всасывается с помощью белка-переносчика, т. е. путём облегчённой диффузии.
Метаболизм фруктозы (рис. 7-69) начинается с реакции фосфорилирования (реакция 1), катализируемой фруктокиназой с образованием фруктозо-1-фосфата. Фермент обнаружен в печени, а также в почках и кишечнике. Этот фермент обладает абсолютной специфичностью, поэтому, в отличие от глюкокиназы, инсулин не влияет на его активность. Последнее обстоятельство объясняет, почему уровень выведения фруктозы в моче у больных сахарным диабетом и здоровых не отличается. Фруктозо-1-фосфат не может превращаться во фруктозо-6-фосфат из-за отсутствия соответствующего фермента. Вместо этого фруктозо-1-фосфат далее расщепляется фруктозо-1-фосфатальдолазой (альдолаза В) на глицеральдегид и дигидроксиацетон- 3-фосфат (реакция 2). Последний является промежуточным продуктом гликолиза и образуется в ходе реакции, катализируемой фруктозо-1,6-бисфосфосфатальдолазой (альдолаза А). Глицеральдегид может включаться в гликолиз после его фосфорилирования с участием АТФ (реакция 3). Две молекулы триозофосфатов либо распадаются по гликолитическому пути, либо конденсируются с образованием фруктозо-1,6- бисфосфата и далее участвуют в глюконеогенезе (реакции 8, 7, 5, 9). Фруктоза в печени включается главным образом во второй путь. Часть дигидроксиацетон-3-фосфата может восстанавливаться до глицерол-3-фосфата и участвовать в синтезе триацилглицеролов.
Рис. 7-69. Метаболизм фруктозы, а — превращение фруктозы в дигидроксиацетон-3-фосфат и глицеральдегид-3-фосфат; б — путь включения фруктозы в гликолиз и глюконеогенез; в — путь включения фруктозы в синтез гликогена.
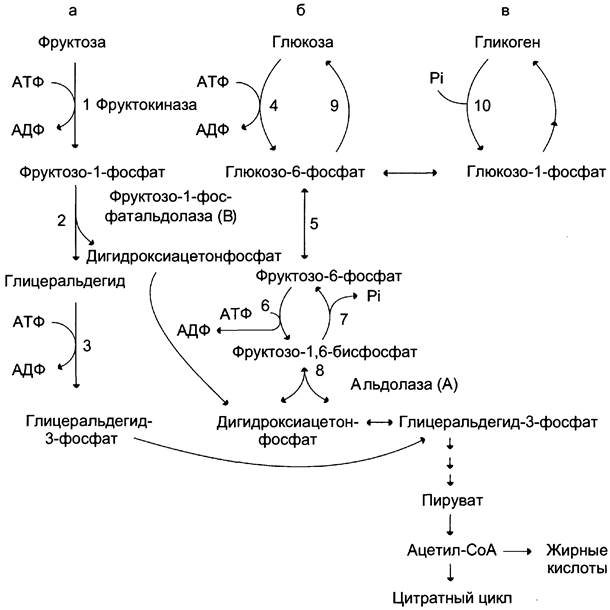
Следует отметить, что включение фруктозы в метаболизм через фруктозо-1-фосфат минует стадию, катализируемую фосфофруктокиназой (реакция 6), которая является пунктом метаболитического контроля скорости катаболизма глюкозы. Этим обстоятельством можно объяснить, почему увеличение количества фруктозы ускоряет в печени процессы, ведущие к синтезу жирных кислот, а также их этерификацию с образованием триацилглицеролов.
Б. Нарушения метаболизма фруктозы
Нарушения метаболизма фруктозы, причиной которых является дефект ферментов, отражены в табл. 7-5.
Таблица 7-5. Нарушения метаболизма фруктозы
Неактивный фермент |
Блокируемая реакция |
Локализация фермента |
Клинические проявления и лабораторные данные |
Фруктокиназа |
Фруктоза + АТФ —> Фруктозо-1-фосфат + АДФ |
Печень Почки Энтероциты |
Фруктоземия, фруктозурия |
Фруктозо-1-фосфатальдолаза |
Фруктозо-1-фосфат —> Дигидроксиацетон-3-фосфат + Глицеральдегид |
Печень |
Рвота, боли в животе, диарея, гипогликемия, Гипофосфатемия, фруктоземия, гиперурикемия, хроническая недостаточность функций печени, почек. |
Недостаточность фруктокиназы клинически не проявляется. Фруктоза накапливается в крови и выделяется с мочой, где её можно обнаружить лабораторными методами. Очень важно не перепутать эту безвредную аномалию с сахарным диабетом. Данное заболевание известно, как доброкачественная эссенциальная фруктозурия и встречается с частотой 1:130 000.
Наследственная непереносимость фруктозы, возникающая при генетически обусловленном дефекте фруктозо-1-фосфатальдолазы, не проявляется, пока ребёнок питается грудным молоком, т. е. пока пища не содержит фруктозы. Симптомы возникают, когда в рацион добавляют фрукты, соки, сахарозу. Рвота, боли в животе, диарея, гипогликемия и даже кома и судороги возникают через 30 мин после приёма пищи, содержащей фруктозу. У маленьких детей и подростков, продолжающих принимать фруктозу, развиваются хронические нарушения функций печени и почек. Непереносимость фруктозы — достаточно частая аутосомно-рецессивная форма патологии.
Дефект альдолазы фруктозо-1 -фосфата сопровождается накоплением фруктозо-1-фосфата, который ингибирует активность фосфоглюкомутазы, превращающей глюкозо-1-фосфат в глюкозо-6-фосфат и обеспечивающей включение продукта гликогенфосфорилазной реакции в метаболизм. Поэтому происходит торможение распада гликогена на стадии образования глюкозо-1-фосфата, в результате чего развивается гипогликемия. Как следствие, ускоряется мобилизация липидов и окисление жирных кислот. Следствием ускорения окисления жирных кислот и синтеза кетоновых тел, замещающих энергетическую функцию глюкозы, может быть метаболический ацидоз (см. раздел 8), так как кетоновые тела являются кислотами и при высоких концентрациях снижают pH крови.
Результатом торможения гликогенолиза и гликолиза является снижение синтеза АТФ. Кроме того, накопление фосфорилированной фруктозы ведёт к нарушению обмена неорганического фосфата и гипофосфатемии.
Для пополнения внутриклеточного фосфата ускоряется распад адениловых нуклеотидов. Продукты распада этих нуклеотидов включаются в катаболизм, проходя стадии образования гипоксантина, ксантина и, наконец, мочевой кислоты. Повышение количества мочевой кислоты и снижение экскреции уратов в условиях метаболического ацидоза проявляются в виде гиперурикемии. Следствием гиперурикемии может быть подагра даже в молодом возрасте (см. раздел 10).
В. Метаболизм галактозы
Галактоза образуется в кишечнике в результате гидролиза лактозы. Чтобы превратить галактозу в глюкозу, необходимо изменить оптическую конфигурацию Н- и ОН-групп С4атома в галактозе, т. е. провести реакцию эпимеризации. Эта реакция в клетке возможна только с УДФ-производным галактозы. УДФ-галактоза образуется из УДФ-глюкозы (метаболит в синтезе гликогена) в ходе реакции, катализируемой уридилфосфат-4- эпимеразой (рис. 7-70, 7-71).
Рис. 7-70. Обмен галактозы.

Рис. 7-71. Реакция эпимеризации УДФ-глюкозы в УДФ-галактозу.

Однако включению галактозы в описанную реакцию эпимеризации предшествует её фосфорилирование с образованием галактозо-1- фосфата (реакция 1 на рис. 7-70). Далее галактозо-1-фосфат замещает остаток глюкозы в УДФ-глюкозе с образованием УДФ-галактозы (реакция 2), т. е. прямая реакция фосфорилированной галактозы с УТФ не происходит.
Реакцию 2 можно рассматривать как перенос уридильного остатка с УДФ-глюкозы на галактозу, поэтому фермент назван галактозо- 1 -фосфатуридилтрансферазой (ГАЛТ).
Затем галактоза в составе нуклеотида включается в реакцию эпимеризации, в которой участвует эпимераза — NAD-зaвиcимый фермент, катализирующий окисление и восстановление галактозы по С4 углеродному атому (реакция 3).
Эпимераза может работать и в другом направлении, преобразуя УДФ-глюкозу в УДФ-галактозу. Эта обратная эпимеризация важна ддя синтеза галактозильных остатков в гликолипидах и гликопротеинах. Кроме того, галактоза необходима для синтеза лактозы в грудных железах. В период лактации галактоза не является незаменимым компонентом пищи, так как может образовываться из глюкозы.
Глюкозо-1-фосфат, образованный в реакции 2, может включаться в разные метаболические пути: 1) синтез гликогена после реакции с УДФ и образования УДФ-глюкозы; 2) превращение в печени в свободную глюкозу и поддержание её концентрации в крови; 3) катаболизм, сопряжённый с синтезом АТФ, и т. д. (см. рис. 7-70).
Г. Нарушения метаболизма галактозы
Обмен галактозы особенно интересен в связи с наследственным заболеванием — галактоземией.
Галактоземия возникает при нарушении обмена галактозы, обусловленном наследственным дефектом любого из трёх ферментов, включающих галактозу в метаболизм глюкозы (табл. 7-6).
Таблица 7-6. Нарушения обмена галактозы
Дефектный фермент (частота) |
Блокируемая реакция |
Клинические проявления и лабораторные данные |
Галактокиназа (1:500 000) |
Галактоза + АТФ —> Галактозо-1 фосфат + АДФ |
Галактоземия, галактозурия, катаракта. Активность фермента в эритроцитах нормальная. |
Галактозо-1 -фосфат- уридилтрансфераза (1:40 000) |
Галактозо-1 фосфат+ УДФ- глюкоза —> УДФ-галактоза + Глюкозо-1 -фосфат |
Галактоземия, галактозурия, галактозо-1 -фосфатемия, катаракта. Тенденция к гипогликемии, компенсаторная мобилизация жиров, цирроз печени, нарушения функции почек. Гепатомегалия, задержка психического развития. Активность фермента в эритроцитах снижена. |
Уридилфосфат-4-эпимераза (1:1 000 000) |
УДФ-глюкоза <-> УДФ-галактоза |
Галактоземия, галактозурия. Тяжёлых клинических проявлений нет. Описаны единичные случаи заболевания. |
Галактоземия, вызванная недостаточностью галактозо-1 -фосфатуридилтрансферазы (ГАЛТ), наиболее хорошо изучена. Это заболевание проявляется очень рано, и особенно опасно для детей, так как основным источником углеводов для них служит материнское молоко, содержащее лактозу. Ранние симптомы дефекта ГАЛТ: рвота, диарея, дегидратация, уменьшение массы тела, желтуха. Они появляются вскоре после рождения, как только ребёнок начинает получать молоко. В крови, моче и тканях повышается концентрация галактозы и галактозо-1-фосфата. В тканях глаза (в хрусталике) галактоза восстанавливается альдоредуктазой с образованием галактитола (дульцита). В этой реакции в качестве донора водорода используется NADPH. Восстановление галактозы происходит и в ходе нормального метаболизма, но протекает с небольшой скоростью. При галактоземии галактитол накапливается в стекловидном теле и связывает большое количество воды. Вследствие этого нарушается баланс электролитов, а чрезмерная гидратация хрусталика приводит к развитию катаракты, которая наблюдается уже через несколько дней после рождения.
Тяжёлые последствия дефекта ГАЛТ наблюдают в печени. Это связано с накоплением галактозо-1-фосфата и его токсическим действием на гепатоциты. В результате возникают нарушения функции печени: гепатомегалия, жировая дистрофия. В почках таких больных также повышена концентрация галактитола и галактозо-1-фосфата, что влияет на их функции. Отмечают нарушения в клетках полушарий головного мозга и мозжечка, в тяжёлых случаях — отёк мозга, задержку умственного развития, возможен летальный исход.
Для галактоземии, вызванной дефектом галактокиназы, тоже характерна катаракта, но при этом заболевании, в отличие от дефекта ГАЛТ, не отмечают нарушений функций печени, почек, мозга. Наиболее тяжёлые последствия снижения активности ГАЛТ связывают с влиянием галактозо-1-фосфата на активность других ферментов, участвующих в углеводном
обмене (фосфоглюкомутазы, глюкозо-6-фос- фатдегидрогеназы).
Известно несколько форм галактоземии, причиной которой является недостаточность ГАЛТ (табл. 7-7).
Таблица 7-7. Некоторые варианты генетического дефекта ГАЛТ
Изменения в структуре ГАЛТ |
Проявления |
Асн —> Асп |
Признак Дюарта. У гетерозигот при этом варианте активность фермента составляет 75% от нормальной. Гомозиготный фенотип Дюарта обычно связан с 50% потерей активности. Пациенты с синдромом Дюарта могут быть здоровыми, несмотря на структурную аномалию ГАЛТ. |
Глн —> Арг |
Проявляется как тяжёлая галактоземия. Причина — мутация типа замены нуклеотида 591 в гене фермента. Активность ГАЛТ составляет 10% от нормы. Эта форма встречается в 70% случаев заболевания галактоземией среди европеоидов, частота — 1:338 886. |
Сер —> Лей |
Заболевание описано у чернокожих пациентов и названо «чёрный признак». Галактоземия проявляется как результат недостаточной активности ГАЛТ в печени и эритроцитах. Активность ГАЛТ в печени составляет 10% от нормы. Тем не менее отмечалась утилизация некоторого количества галактозы, что объяснялось развитием альтернативного пути. Причина — мутация типа замены 1158-го нуклеотида в гене фермента. |
Арг —> Три |
Тяжёлая форма галактоземии. Причина — миссенс-мутация нуклеотида 1025 в гене фермента. Активность ГАЛТ отсутствует. |
Лиз —> Асн |
Широко распространённая мутация при галактоземии. |
Некоторые дефекты в строении ГАЛТ приводят лишь к частичной потере активности фермента. Поскольку в норме ГАЛТ присутствует в организме в избытке, то снижение его активности до 50%, а иногда и ниже может клинически не проявляться.
При диагностике галактоземии исследуют мочу на содержание галактозы, собранную после нескольких кормлений молоком. При обнаружении у ребёнка катаракты его обследуют на недостаточность галактокиназы и ГАЛТ. Наличие галактозы в моче при отсутствии нарушений функции печени указывает на дефект галактокиназы. При обследовании проведение теста с нагрузкой галактозой не рекомендуется, так как этот тест опасен для больных. Лечение заключается в удалении галактозы из рациона.